-
Looking for chiral recognition in photoinduced bimolecular electron transfer using ultrafast spectroscopy
P. Verma, C. Nancoz, J. Bosson, G.M. Labrador, J. Lacour and E Vauthey
Physical Chemistry Chemical Physics, 25 (2023), p11111-11120


DOI:10.1039/D3CP00760J | unige:168393 | Abstract | Article HTML | Article PDF | Supporting Info
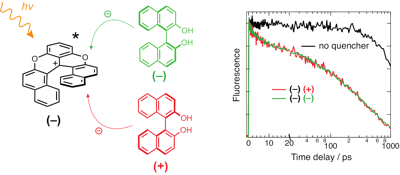
Occurrence of chiral recognition in bimolecular photoinduced electron transfer (ET) is difficult to identify because of the predominant role of diffusion. To circumvent this problem, we apply a combination of ultrafast time-resolved fluorescence and transient electronic absorption to look for stereoselectivity in the initial, static stage of ET quenching, where diffusion is not relevant. The fluorophore and electron acceptor is a cationic hexahelicene, whereas the quencher has either stereocentered (tryptophan) or axial (binaphthol) chirality. We found that, in all cases, the quenching dynamics are the same, within the limit of error, for different diastereomeric pairs in polar and medium-polar solvents. The same absence of chiral effect is observed for the recombination of the radical pair, which results from the quenching. Molecular dynamics simulations suggest that the distribution of inter-reactant distance is independent of the chirality of the acceptor and the donor. Close contact resulting in large electronic coupling is predicted to be possible with all diastereomeric pairs. In this case, ET is an adiabatic process, whose dynamics do no longer depend on the coupling, but are rather controlled by high-frequency intramolecular modes.